What are the molecular mechanisms that underlie neuronal degeneration? Do mechanisms of prion toxicity differ depending on the prion conformation? Which cells contribute to the synapse loss and neuronal death?
In addition to prions, amyloid-β and α-synuclein aggregates also bind to the cellular prion protein, triggering signaling cascades that accelerate neuronal degeneration. We have developed a new transgenic mouse model by CRISPR-Cas that shows hippocampal neuronal pathology similar to that in prion disease. Ultrastructurally, there are dystrophic neurites, indicating a failure of protein clearance mechanisms in neurons, as seen in the brains of patients with prion or Alzheimer’s disease.
A research goal is to interrogate the cellular pathways altered in the initial prion disease states using simple to complex disease models. We are currently using models with deficiencies in different cellular aggregate processing pathways to better understand the points of failure in protein aggregate clearance. Using a variety of prion strains and super-resolution confocal microscopy, we are currently working to identify the cellular factors that facilitate or hinder disease progression in order to develop therapies that prevent long-term damage to neurons.
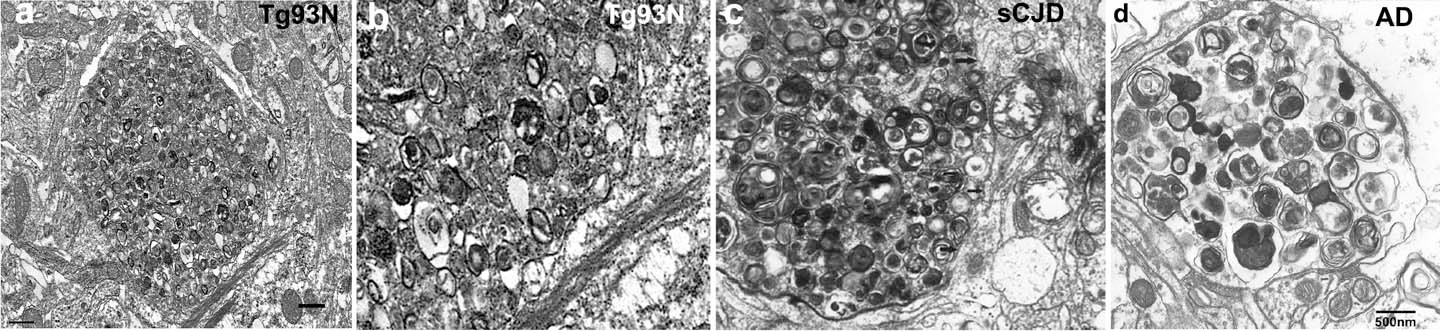
Ultrastructure of hippocampus shows a dystrophic neurite filled with autophagic vacuoles
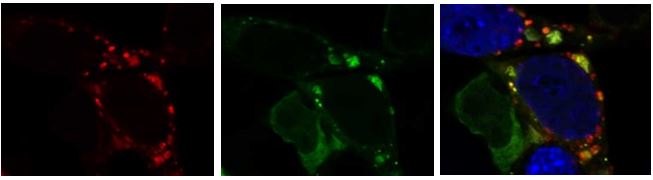
N2a neuroblastoma cell transfected with mCherry-GFP-tagged LC3 tagged tandem reporter shows the amphisomes or autolysosomes in red only (arrows), and phagophores or autophagosomes (not yet fused with the lysosome) where green and red co-localize (the GFP is degraded in the low pH conditions of the lysosome). Image collected using a Zeiss 880 airyscan super resolution confocal microscope in the UCSD microscopy core.